Introduction
Modern genetic association studies routinely employ data on tens to hundreds of thousands of individuals, genotyped or imputed at tens of millions of markers genome-wide. Traditional data formats based on text representation of these data - such as the GEN format output by IMPUTE, or the Variant Call Format - are sometimes not well suited to these data quantities. Indeed, for simple programs the time spent parsing these formats can dominate program execution time.
This page describes a binary GEN file format (the "BGEN" format) which aims to address these problems. BGEN is a robust format that has been designed to have a specific blend of features that we believe make it useful for this type of study. It is targetted for use with large, potentially imputed genetic datasets. Key features include:
For example, the following plot shows the time taken to list variant identifying data - i.e. the genomic position, ID fields and alleles - for various common formats (Y-axis), against file size (X axis), for a dataset of 18,496 samples typed at 121,668 SNPs on chromosome 1. Both variants of BGEN defined below are shown.
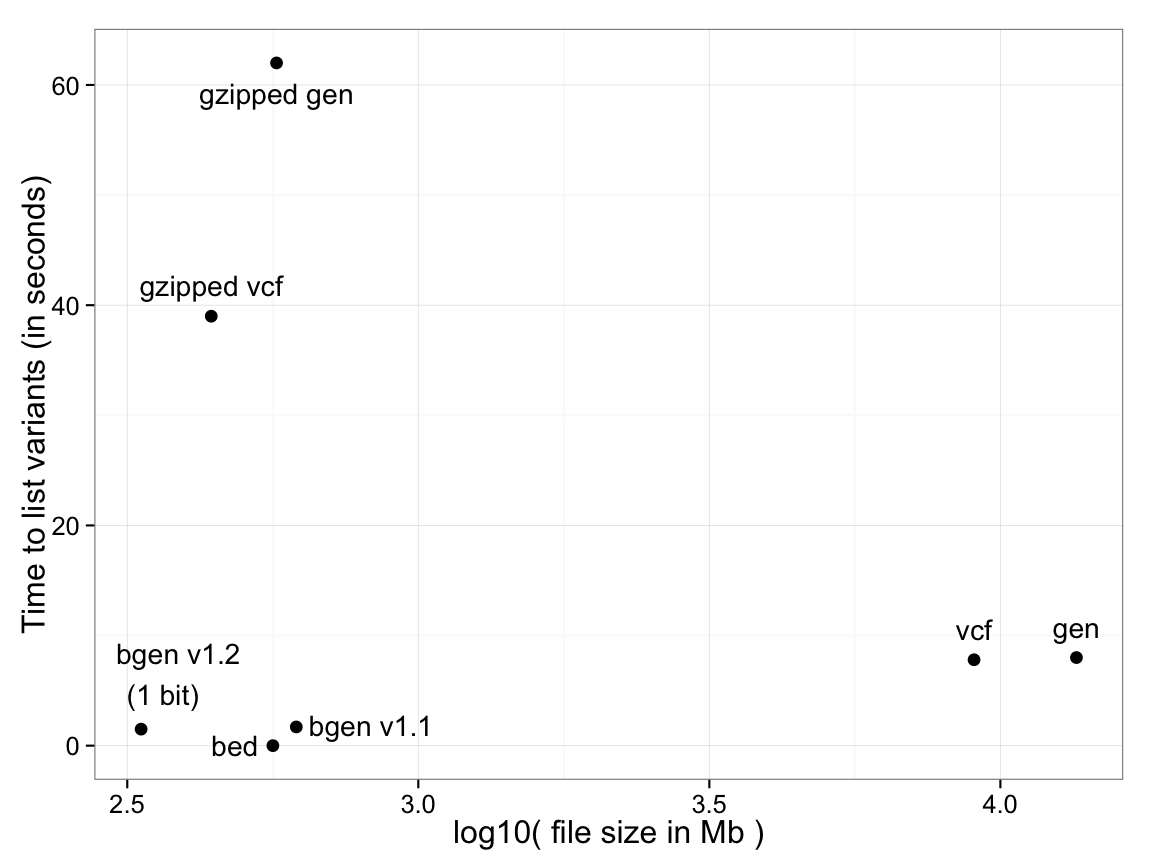
For PLINK binary (.bed) files, identifying data is
stored in a separate file (the .bim file) so the time is effectively zero.
For text-based formats there is a significant trade-off between the use of file compression and
read performance. BGEN stores
the entire dataset of 2,250 million genotypes in 334Mb, slightly over one bit per genotype, and in this test took 1.5s.
(Performance optimisation of all formats may of course be possible, so the above plot will not represent the best possible timings, but should be regarded as illustrative.)
The BGEN format has been used in several major projects, including the Wellcome Trust Case-Control Consortium 2, the MalariaGEN project, and the ALSPAC study. It has been adopted as the release format for genome-wide imputed genotypes for the UK Biobank.